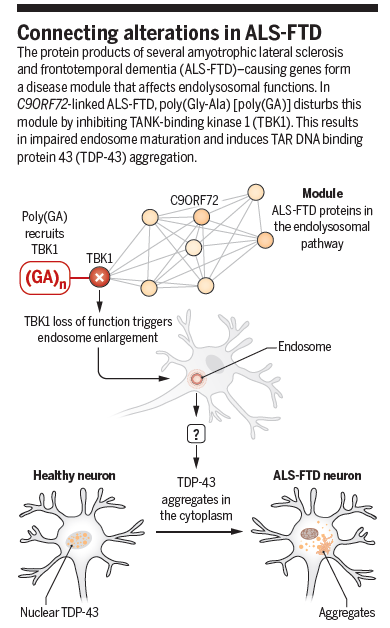
额颞叶痴呆(Frontotemporal dementia,FTD)和肌萎缩性侧索硬化症(Amyotrophic lateral sclerosis,ALS)与C9orf72基因的重复扩增和TBK1(TANK-binding kinase 1)基因的突变有关。有研究发现部分患者同时存在C9orf72重复扩增与TBK1的突变,这些患者会出现发病年龄提早以及病程发展变快的情况。因此,TBK1可能调节C9orf72相关的FTD-ALS的发生和发展。为了研究这两个基因在FTD-ALS中的作用,美国梅奥医学中心Leonard Petrucelli/Yong-Jie Zhang团队(第一作者:邵炜博士)在Science发文题为Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration,TBK1在C9orf72中重复多聚化时被磷酸化,随后包裹进入聚集体(Aggregates)中导致TBK1活性的丧失,最终导致神经退行性变。
额颞叶痴呆以及脊髓侧索硬化症最主要的特征是形成蛋白质聚集体,特别是TDP-43会形成聚集体。聚集体的形成意味着蛋白质清除受损,这些FTD-ALS相关的基因影响蛋白质稳态。为了检测TBK1突变以及C9orf72中GC重复对于FTD-ALS的影响,作者们使用基于腺相关病毒的C9orf72-(G4C2)149小鼠模型,其中C9orf72中GC达到149个重复,从而确定C9orf72的重复扩增是否影响TBK1磷酸化状态。作者们发现C9orf72-(G4C2)149小鼠和C9orf72-(G4C2) 2对照组的大脑皮层裂解液中TBK1总水平相当,但是pTBK1的水平在C9orf72-(G4C2)149小鼠中显著增加。另外,C9orf72-(G4C2)149与pTBK1共同定位在聚集体中。因此,C9orf72-(G4C2)149能够触发小鼠脑内TBK1的磷酸化以及异常聚集(图1)。
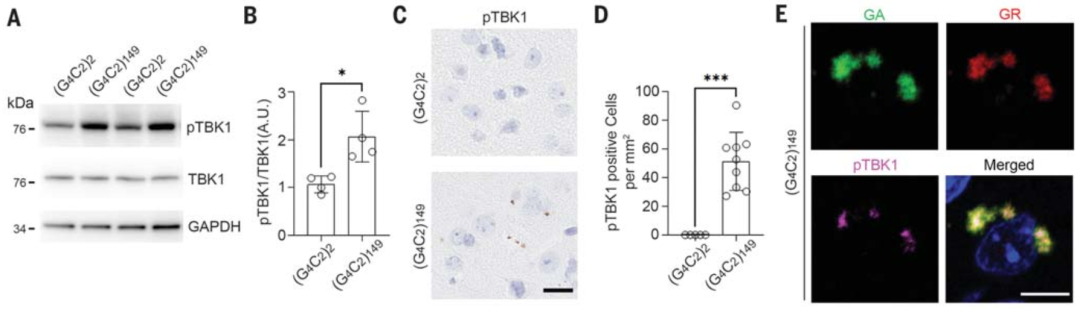
图1 TBK1在C9orf72中GC重复扩增后磷酸化增加并被隔离到包裹体中
作者们认为C9orf72-(G4C2)149的聚集体会将pTBK1进行隔离,TBK1从而没有办法发挥调节聚集体自噬清除的功能。作者们发现聚集体表达的细胞中表达野生型TBK1时会导致自噬受体蛋白p62磷酸化的显著增加。但是在过表达自磷酸化活性降低的部分功能丧失突变型TBK1时,聚集体则不会出现减少。因此,TBK1被聚集体隔离,将会损害TBK1的活性,加剧神经元缺陷。通过将TBK1突变型以及聚集体同时表达的小鼠同时敲入来模拟双突变情况,作者们发现会导致出现神经退行性疾病特征比如总脑重量减少、皮质神经元丢失等。
TBK1丢失会损伤人类运动神经元内体成熟(Endosome maturation)过程。作者们想知道TBK1的隔离是否也会影响体内内体成熟信号通路。作者们发现聚集体表达的小鼠皮层中早期内体会出现增大的表型,同时Rab7阳性的晚期内体也会出现异常。另外,作者们发现内体成熟信号通路的异常会导致皮层神经元中TDP-43的病理性特征。由此建立起聚集体、内体缺陷以及TDP-43病例特征之间的联系。
总的来说,作者们的工作发现TBK1在C9orf72重复序列多聚化时磷酸化水平增加,并被隔离到聚集体中,导致TBK1活性的丧失,聚集体增加抑制内体途径诱导TDP-43聚集,进而导致神经退行性变。该工作将C9orf72、TBK1和TDP-43之间的相互作用将FTD-ALS的三个不同方面连接成一个连贯的通路。
同期Science配发了专门评论:A perturbed network in neurodegeneration。评论认为:迄今为止,有超过20个彼此似乎毫无关联的基因被确认为ALS-FTD的致病基因或者重要的调节者;而这项研究建立ALS-FTD重要的三个基因C9orf72、TBK1和TDP-43在内吞-溶酶体路径的互作和影响,为我们理解ALS-FTD的致病机制带来了概念的革新,为疾病提示了新的治疗靶点。
原文链接:http://doi.org/10.1126/science.abq7860